مرضى فقر الدم المنجلي.. معاناة مستمرة وأمل في العلاج الجيني







أنجبت ”فداء“ و”علي“ -36 سنة - طفلهما الأول وكان سليماً من ”المنجلية“، ولكن المرض باغت طفلهما الثاني ”فراس“ - 4 سنوات -، ثم أنجبت الثالث وهي في ريب وقلق من أن يكون هو الآخر مصاباً.
”فراس“ وُلد مصاباً بفقر الدم المنجلي بنسبة 60%، لم تكتشف ”فداء“ مرض طفلها إلا صدفة في إحدى عيادات الأطفال الخاصة التي تتردد عليها بشكل روتيني حينما لاحظت انخفاض مناعة ”فراس“، وكان عمره سنتين ونصف.
بعد أن عرفت ”فداء“ بمرض طفلها ”فراس“ كانت أولى نوبات الألم صادمة بالنسبة لها، فحينما أخذته لمستشفى خاص بسبب حمى تم تنويمه لمدة 8 أيام، ولم يستطيعوا خفض حرارته خلالها بالمضادات الحيوية أو معرفة السبب، وحين نقلته للمستشفى الحكومي وهو مشخص بإصابته بالأنيميا المنجلية، اكتشفوا أنه كان قد انتفخ طحال ”فراس“ واضطروا لنقل الدم له.
”أصبحت أخاف على ”فراس“ من كل شيء، أهتم بطريقة ملبسه، وأبحث عن أي مصدر بُقُولي في كل طعام يتناوله، حتى حين أبعثه للروضة أرفق تقريراً شاملاً بمرضه وما تتطلبه رعايته الصحية وللتواصل معي في أي طارئ“.. وتكمل ”فداء“ بحسرة: ”عندما أرى معاناته ألوم نفسي، رغم أننا في زمن متطور، تتوفر فيه فحوصات الدم والاستشارات في كل مكان، إلا أن التوعية لم تكن كافية، لم يخبرنا والدانا أننا نحمل هذا المرض، أو احتمالية أن أشعر بالألم في أي جزء من جسمي بسببه، ولم نكن مضطرين لعمل الفحوصات إلا من أجل الزواج“.
”زواجنا كان تقليدياً، لم أكن أعرفه أو يعرفني، وحين ظهرت نتيجة برنامج فحص ما قبل الزواج، وكانت النتيجة غير متوافقة، أصر ”علي“ على الزواج بي رغماً عن ذلك، وعلى ذلك وقعنا التعهد بتحمل مسؤولية القرار، في حال أنجبنا أطفالاً مصابين بالمرض“. وتسترسل في حديثها: ”كنت في وقتها لا تنتابني أعراض فقر الدم المنجلي، لم أعان من أي ألم نهائياً، ولذلك لم أكن أعرف أنني حاملة لسمة الأنيميا المنجلية، وكان ”علي“ مثلي كذلك، كنا جاهلين حتى بمقدار نسبته في كل منا، غير أننا علمنا من برنامج الفحص بأننا حاملا سمة المرض.
وقالت وهي تتألم: ”لو يعود الزمن بي، لا أوافق على إتمام الزواج، ليتني كنت واعية بشكل كاف، أنا اليوم أشعر بغصة حين يأتي ”فراس“ يطلب مني أن أسمح له بتناول طعام لا يناسب مصابي“ المنجلية ”، وأحاول جاهدة أن أجعله واعياً لضرره عليه، واعياً بشكل كاف بهذا المرض“ مؤكدة أن زوجها يشاركها ذات الهموم.
وتمتنع ”فداء“ بعد معاناتها مع مرض طفلها، عن نية الإنجاب في المستقبل، حتى لا يتكرر هذا المصير ويورثانه لابن آخر، آملين أن تخفف العلاجات الجديدة من ألم ”فراس“.
 يعد مرض فقر الدم المنجلي مرضاً وراثياً وهو أحد أكثر أمراض الدم الوراثية شيوعًا في العالم، ويحمل ما يقارب 27 مليون مصاب بفقر الدم المنجلي حول العالم، ويحمل 20% سمة فقر الدم المنجلي، في حين يولد 300 ألف طفل بهذا المرض كل عام، وذلك بحسب الأستاذ زكريا الكاظم عضو ورئيس المنظمة الدولية لصوت الخلية المنجلية.
يعد مرض فقر الدم المنجلي مرضاً وراثياً وهو أحد أكثر أمراض الدم الوراثية شيوعًا في العالم، ويحمل ما يقارب 27 مليون مصاب بفقر الدم المنجلي حول العالم، ويحمل 20% سمة فقر الدم المنجلي، في حين يولد 300 ألف طفل بهذا المرض كل عام، وذلك بحسب الأستاذ زكريا الكاظم عضو ورئيس المنظمة الدولية لصوت الخلية المنجلية.
يحمل 180 ألف فرد في الشرق الأوسط وشمال أفريقيا سمة الأنيميا المنجلية، وتتصدر المملكة العربية السعودية دول الخليج العربي في عدد الإصابات بنحو 64 ألف شخص، علاوة على أنه أكثر الأمراض الوراثية انتشارا في المنطقة الشرقية بنسبة تتركز فيها تصل إلى 17% من الحاملين للسمة المنجلية، وترصد محافظة القطيف منها سنويا عشرات الوفيات؛ بسبب مضاعفات هذا المرض، في حين تم مؤخراً افتتاح مستشفى متخصص يقدم الرعاية الصحية لهذه الفئة وهو مستشفى الأمير محمد بن فهد لأمراض الدم الوراثية وهو الأول من نوعه في المنطقة بعد أن كان مستشفى القطيف المركزي يلقى ضغطاً في الكفاية السريرية لهذه الفئة.
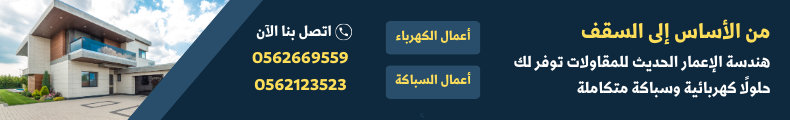
تم الإبلاغ عن مرض فقر الدم المنجلي في المملكة العربية السعودية لأول مرة في المنطقة الشرقية في ستينيات القرن العشرين حيث كانت نسبة انتشاره 25% بين العاملين وذلك بواسطة دراسة استقصائية قامت بها شركة الزيت العربية الأمريكية ”أرامكو“ في المنطقة الشرقية. وقد أدى ذلك إلى بدء دراسات فحص إقليمية ووطنية متعددة لتحديد الخصائص السريرية وتواتر جينات مرض فقر الدم المنجلي في مناطق مختلفة من المملكة.
قدّر برنامج فحص ما قبل الزواج معدل انتشار جين الخلية المنجلية بين السكان البالغين بنسبة 4.2% لسمة الخلية المنجلية و 0.26% لمرض فقر الدم المنجلي، مع تسجيل أعلى معدل انتشار في المنطقة الشرقية ”حوالي 17% لسمة الخلية المنجلية ومرض فقر الدم المنجلي“ 1.2% لمرض فقر الدم المنجلي".
وفي تجربة إقليمية لفحص حديثي الولادة لمرض فقر الدم المنجلي في المنطقة الشرقية على مدار فترة 9 سنوات منذ بدء البرنامج، بلغ معدل انتشار سمة الخلايا المنجلية حوالي 21% وكان معدل انتشار مرض فقر الدم المنجلي 2.6% ”مقارنة بـ 17% و 1.2%، على التوالي، من حالات فحص ما قبل الزواج“.
”فداء“ وزوجها يحملان ”سمة“ فقر الدم المنجلي، وكل منهما يسميان محلياً ”حامل“ للمرض، فتجد أن الأشخاص الذين يحملون سمة الخلية المنجلية لديهم جين هيموغلوبين طبيعي ”HbA“ واحد وجين هيموغلوبين منجلي ”HbS“ واحد. يستطيع جين الهيموغلوبين المنجلي ”HbS“ على تغيير شكل خلايا الدم الحمراء إلى الشكل المنجلي. ولا يعني وجود سمة الخلية المنجلية لدى أحد الأشخاص أنه ”مصاب“ بداء فقر الدم المنجلي. العكس صحيح، بالفعل. فإن سمة الخلية المنجلية منفصلة تمامًا عن المرض؛ وقد تسبب حدوث أعراض، إلا أن حالات حدوث ذلك تُعد نادرة.
وتعتمد نسبة شدة مرض الأنيميا المنجلية على عدد خلايا الدم الحمراء المنجلية الموجودة في الجسم. فكلما زاد عدد هذه الخلايا، زادت شدة المرض، ويتم تحديدها عن طريق تحليل الدم، حيث يتم قياس نسبة خلايا الدم الحمراء المنجلية إلى إجمالي عدد خلايا الدم الحمراء. وتكون شدة الأنيميا المنجلية حسب نسبة خلايا الدم الحمراء المنجلية هي: أقل من 30%: خفيفة، 30-60%: متوسطة. أكثر من 60%: شديدة.
وتتضح نسبة شدة المرض في أي عمر، ولكن عادة ما يتم تشخيص المرض في مرحلة الطفولة. وعبر ذلك يتم تمييز حامل المرض من المصاب بها من خلال تحليل الدم. فحامل المرض لديه نسخة واحدة من الجين المسبب للمرض، بينما المصاب لديه نسختان من الجين، وقد تظهر عليه الأعراض، مثل: نوبات الألم، فقر الدم، الالتهابات، تلف الأعضاء.
وشدة المرض يمكن أن تختلف من شخص لآخر، حتى لو كانت النسبة متشابهة. فهناك بعض الأشخاص المصابين بالمرض بأعراض شديدة، بينما هناك أشخاص آخرون مصابين بالمرض بأعراض خفيفة، وهناك بعض العوامل التي يمكن أن تؤثر على شدة المرض، مثل: العمر، الجنس، البيئة، العلاجات الطبية.
فتنقسم شدة الأنيميا المنجلية إلى ثلاث مراحل، وتعتبر شدة خفيفة حين يكون عدد خلايا الدم الحمراء المنجلية أقل من 30% من إجمالي عدد خلايا الدم الحمراء. عادة ما تكون الأعراض خفيفة أو غائبة، وقد يعاني الشخص من بعض الألم أو فقر الدم.
بينما تكون شدة متوسطة حين يكون عدد خلايا الدم الحمراء المنجلية بين 30 و 60% من إجمالي عدد خلايا الدم الحمراء. قد تشمل الأعراض نوبات الألم الشديدة، وفقر الدم المزمن، والالتهابات المتكررة. وحين تكون شدة شديدة: يكون عدد خلايا الدم الحمراء المنجلية أكثر من 60% من إجمالي عدد خلايا الدم الحمراء وهنا قد تشمل الأعراض نوبات الألم الشديدة، وفقر الدم المزمن، والالتهابات المتكررة، وتلف الأعضاء.
عندما يتزوج الحاملون لسمة فقر الدم المنجلي، فإن نسبة أن ينجبوا أطفالاً سليمين هي 25%، بينما احتمالية حمل السمة تكون بنسبة 50%، في حين أن الإصابة بفقر الدم المنجلي ستكون بنسبة 25%، والمصابون بفقر الدم المنجلي معرضون لحدوث نوبات دورية من الألم الشديد، تسمى نوبات الألم. ويحدث الألم عندما تمنع خلايا الدم الحمراء المنجلية تدفق الدم عبر الأوعية الدموية الصغيرة إلى الصدر والبطن والمفاصل. يختلف الألم في شدته، وقد يستمر لبضع ساعات أو أيام، فتحدث نوبة الألم لطفل عمره 8 سنوات تمتد لنحو 32 مرة في العام الواحد، وتستمر من 5 - 21 يومًا وذلك بحسب زكريا الكاظم رئيس المنظمة الدولية لصوت الخلية المنجلية.
ويحدث لدى معظم المصابين بفقر الدم المنجلي ضخامة في الطحال في أثناء الطفولة - وهو العضو المسؤول عن مقاومة العدوى، وينقي الدم من المواد غير الضرورية مثل خلايا الدم القديمة أو التالفة. - لأن الخلايا المنجلية تصبح محتجزة في الطحال. وفي الوقت الذي يصل فيه الشخص إلى سن المراهقة، يكون الطحال قد تضرر كثيراً في أغلب الأحيان، بحيث يصبح منكمشا وفاقدا لوظائفه، لأنه يتلاشى ذاتيا بمرور الوقت إذ أنه تعرض لحدوث جلطات متتابعة تؤدي إلى ضموره وصغر حجمه وهذا يسمى ”الاستئصال الذاتي للطحال“.
وبما أن الطحال يساعد على مكافحة العدوى، لذلك يكون المصابون بداء الخلية المنجلية أكثر ميلاً للإصابة بالالتهاب الرئوي أو متلازمة الصدر الحادة، والتي تعد من المضاعفات التي تهدد الحياة، ويمكن أن تؤدي إلى إصابة الرئة، وصعوبة التنفس، وانخفاض الأكسجين لبقية الجسم. أو يتعرضون لموت أنسجة العظام، تجلط الدم، مشاكل الكلى، تقرحات الساق، مشاكل في الكبد، السكتة الدماغية، أو فقدان البصر.
ففي دراسة ابتدائية نشرها فريق طبي سعودي في المجلة البريطانية لأمراض الدم عام 2014 لتقييم نمط مضاعفات مرض فقر الدم المنجلي لدى البالغين. تم تسجيل ما مجموعه 104 مريضا بمتوسط عمر 27 عاما. أبلغ 96% من هؤلاء المرضى عن تاريخ من الأزمات المؤلمة. 47% أصيبوا بنوبة واحدة على الأقل من متلازمة الصدر الحادة، ومع ذلك، 15% فقط أصيبوا بنوبتين أو أكثر. تم الإبلاغ عن نخر العظم المصحوب بأعراض في 18%؛ القساح بنسبة 17%؛ السكتة الدماغية العلنية بنسبة 6%؛ لم يكن أي منهم يعاني من تقرحات في الساق. وكان غالبية المرضى يعانون من تضخم الطحال المستمر، بينما 66% منهم يعانون من حصوات في المرارة. نصف المرضى شاركوا في وراثة ثلاسيميا ألفا.
ويعد ثلاسيميا ألفا اضطراب في الدم يقلل من إنتاج الهيموغلوبين الطبيعي وهو المسؤول عن حمل الأكسجين إلى الخلايا في جميع أنحاء الجسم، وله عدة أنواع مختلفة تتراوح في حدتها بين الطفيف والشديد، فبينما قد لا تسبب بعض أنواع الثلاسيميا أكثر من فقر دم خفيف الحدة، قد يكون بعضها الآخر قاتلًا ليسبب وفاة الجنين المصاب قبل ولادته، أو بعد ولادته بفترة.
 وقال الدكتور عبد الله آل زايد استشاري أمراض الدم ورئيس مركز أمراض الدم الوراثية بمستشفى القطيف المركزي من ناحية الأدوية والعلاجات: ”بقي الاهتمام بعلاجات هذا المرض متواضعا لأكثر من عقدين من الزمن، وكان آخر علاج معترف به هو علاج“ الهيدروكسي يوريا ”في 1998 م وهو أكثر علاج فعال متاح حالياً، ثم توالت الأدوية الحديثة في الظهور في السنوات الثلاث الماضية“.
وقال الدكتور عبد الله آل زايد استشاري أمراض الدم ورئيس مركز أمراض الدم الوراثية بمستشفى القطيف المركزي من ناحية الأدوية والعلاجات: ”بقي الاهتمام بعلاجات هذا المرض متواضعا لأكثر من عقدين من الزمن، وكان آخر علاج معترف به هو علاج“ الهيدروكسي يوريا ”في 1998 م وهو أكثر علاج فعال متاح حالياً، ثم توالت الأدوية الحديثة في الظهور في السنوات الثلاث الماضية“.
ولفت إلى ظهور ثلاثة أدوية جديدة حصل واحد منها على موافقة هيئة الغذاء والدواء السعودية، وهو علاج ال ”كريزنليزوماب“، وهناك دواء آخر قاب قوسين من أخذ الموافقة من هيئة الغذاء والدواء، وهو علاج ”إل - جلوتامين“، أما العلاج الثالث فهو ”فوكسلوتور“.
وقال الدكتور عبدالله: ”لأول مرة منذ سنوات نرى اهتماماً غير مسبوق في البحث عن علاجات جديدة لمرضى فقر الدم المنجلي“.
ولفت إلى أن هذه العلاجات تهدف إلى السيطرة على المرض وليس الشفاء منه، وتبقى زراعة الخلايا الجذعية أو زراعة نخاع العظم هو العلاج الوحيد المعتمد للشفاء من المرض، ويفضل أن تكون الزراعة قبل عمر ستة عشر عاماً لتكون النتائج أفضل، ولكن قد تتم الزراعة بعد هذا العمر وبنتائج جيدة جداً.
بدوره، كشف آل زايد مؤخراً في خبر نشرته ”جهات الإخبارية“ عن موافقة وكالة تنظيم الأدوية ومنتجات الرعاية الصحية ”MHRA“ في المملكة المتحدة على العلاج الجيني الجديد لمرضى فقر الدم المنجلي ومرضى الثلاسيميا بيتا المعتمدين على نقل الدم.
حيث كشفت الوكالة أن العقار ”كاسجيفي“ ”Exagamglogene autotemcel“ يعد أول دواء في العالم يعتمد أداة تعديل الجينات المبتكرة ”كريسبر“ ”CRISPR“، التي فاز مخترعاها بجائزة نوبل عام 2020.
وتم تصميم ”كاسجيفي“ للعمل عن طريق تعديل الجين المعيب في الخلايا الجذعية لنخاع عظم المريض بحيث ينتج الجسم الهيموجلوبين الوظيفي. وللقيام بذلك، يتم أخذ الخلايا الجذعية من نخاع العظم، وتحريرها في المختبر ومن ثم غرسها مرة أخرى في المريض، وبعد ذلك من المرجح أن تستمر النتائج مدى الحياة.
وقال: ”إن العلاج يستهدف المرضى من عمر 12 عاما فما فوق، وهذا الاعتماد الأول من بريطانيا، ولكننا نترقب الاعتماد الأهم من هيئة الغذاء والدواء الأمريكية والمتوقع الإعلان عنه في شهر ديسمبر القادم، أي تفصلنا عنه أسابيع قليلة فقط“.
وأوضح أن العلاج شبيه بزراعة الخلايا الجذعية الذاتية، وللقيام بذلك، يتم أخذ الخلايا الجذعية من نخاع العظم من المريض نفسه، ويتم تحريرها وتعديلها في المختبر، ومن ثم غرسها مرة أخرى في المريض، معولاً بقوله: ”نأمل أن تستمر النتائج مدى الحياة“.
 تواجه المملكة العربية السعودية تحديات عديدة في السيطرة على مرض فقر الدم المنجلي. أحد التحديات الرئيسية هو انتشار زواج الأقارب، ويمكن أن يرتفع هذا الانتشار في القرى أو بين القبائل. وبما أن هذه الزيجات تزيد من خطر الإصابة بالأمراض الوراثية، تحاول الحكومة السعودية توعية الناس بآثار زواج الأقارب من خلال برامج التوعية. وأنجح البرامج التي نفذتها الحكومة هي برنامج فحص ما قبل الزواج وبرنامج الاستشارة الوراثية. يعد برنامج فحص ما قبل الزواج إلزاميًا لجميع الأزواج، ويتم تقديمه مجانًا في جميع المرافق الحكومية. ويبدو أن هذا البرنامج ناجح، حيث نجح بشكل كبير في خفض عدد حالات الإلغاء الطوعي لعروض الزواج المعرضة للخطر.
تواجه المملكة العربية السعودية تحديات عديدة في السيطرة على مرض فقر الدم المنجلي. أحد التحديات الرئيسية هو انتشار زواج الأقارب، ويمكن أن يرتفع هذا الانتشار في القرى أو بين القبائل. وبما أن هذه الزيجات تزيد من خطر الإصابة بالأمراض الوراثية، تحاول الحكومة السعودية توعية الناس بآثار زواج الأقارب من خلال برامج التوعية. وأنجح البرامج التي نفذتها الحكومة هي برنامج فحص ما قبل الزواج وبرنامج الاستشارة الوراثية. يعد برنامج فحص ما قبل الزواج إلزاميًا لجميع الأزواج، ويتم تقديمه مجانًا في جميع المرافق الحكومية. ويبدو أن هذا البرنامج ناجح، حيث نجح بشكل كبير في خفض عدد حالات الإلغاء الطوعي لعروض الزواج المعرضة للخطر.
ومن جانبه، أشار الدكتور زهير رهبيني استشاري أمراض الوراثة وطب الأطفال وعضو مجلس إدارة فحص ما قبل الزواج لمرضى الأنيميا المنجلية وأنيميا البحر الأبيض المتوسط في العام 2004. تلاه برنامج الفحص المبكر للمواليد والذي بدأ عام 2005، والذي يكشف اليوم عن 17 مرضاً وراثياً.
يقول رهبيني إنه منذ بداية صدور المرسوم الملكي عام 2004 بضرورة الفحص الطبي ما قبل الزواج فيما يخص مرض الأنيميا المنجلية، وأنيميا البحر المتوسط كان هناك 90% من الأشخاص المقبلين على الزواج يعلمون بحملهم لأحد هذين المرضين، ورغم ذلك أقدموا على الزواج. وخلال الأعوام القريبة الماضية أوضحت الإحصائيات أن نسبة من يعلمون أنهم حاملون للمورثة وأقدموا على الزواج انخفضت إلى أقل من 40%.
وفي دراسة استقصائية عالمية قد يؤثر داء فقر الدم المنجلي ونوبات الألم على العمل والدراسة والحياة الاجتماعية للمصابين، فهنالك شخص واحد من بين 6 أشخاص يتغيب عن العمل بسبب نوبات الألم، بينما يتغيب بعض الأطفال المصابين أكثر من 3 أيام عن المدرسة أو غيرها من النشطة بعد خروجهم من قسم الطوارئ أو المستشفى، كما أن مرض فقر الدم المنجلي قد يكون سبباً للعزلة بسبب البقاء لفترات طويلة في المنزل أو المستشفى.
ويقول ”زكريا الكاظم“: ”لقد وجدنا أن من يحمل سمة المرض يواجه تحديات تصل إلى الموت، وغالبية من سقط في الملاعب أثناء أداء الرياضة ذات جهد بدني عال توفوا نتيجة حملهم سمة المرض، وقد استطعنا أن نخلق تشريعات حول أهمية فحص اللاعبين وعدم تعريض الأفراد الذين يعيشون مع سمة المرض إلى أي رياضات فيها إجهاد بدني أو حراري، وقد استجابت الولايات المتحدة كما واستجابت مملكة البحرين في سن قانون يحد من انخراط أو تعريض الأفراد الذين يعيشون مع سمة المرض إلى هذه الرياضات، بل ذهبت الولايات المتحدة إلى إعفاء حاملي سمة المرض من التجنيد الإجباري“.
من جانبه، قال المختص الاجتماعي جعفر العيد: ”إن مرض فقر الدم المنجلي يؤثر على الأسرة والمجتمع بشكل كبير، إذ يشكل عبئاً مالياً على الأسرة، حيث يحتاج المريض إلى رعاية طبية مستمرة. كما أن المرض قد يتسبب في إعاقة المريض، مما يؤثر على قدرته على العمل والإنتاج، كما قد يتسبب في تغيب متكرر للمريض عن العمل، مما قد يؤثر على سمعته المهنية.“.
وأوضح أن المرض قد يؤثر على العلاقات الاجتماعية للمريض، حيث قد يشعر المريض بالعزلة والوحدة بسبب مرضه. وقال العيد: ”مرض فقر الدم المنجلي هو مشكلة اجتماعية واقتصادية. يجب أن تتضافر الجهود لتوفير الدعم للمصابين بالمرض ولعائلاتهم“.
وأوضح أن المصابين بمرض فقر الدم المنجلي قد يواجهون صعوبة في الحصول على عمل، حيث قد يتردد أصحاب العمل في توظيفهم بسبب مرضهم.
تضرر ”أحمد“ رغم عدم درايته وآخرين كثر، عن وجود إحدى مواد نظام العمل المعمول به ضمن المملكة العربية السعودية، والخاص بتحديد شروط وضوابط عمليات التوظيف في السعودية، ووقاية العامل من مخاطر العمل، وتوفير الخدمات الاجتماعية والصحية لجميع العمال، وذلك ما تنص عليه المادة رقم 117 من نِظام العمل السعودي.
حيث المادة وضعت وحددت الأجر المستحق للعامل عبر 120 يوم إجازة مرضية، ونصها: ”للعامل الذي يثبت مرضه الحق في إجازة مرضية بأجر عن 30 يومًا الأولى، و 75% من الأجر عن 60 يومًا التالية، ودون أجر 30 يومًا التي تلي ذلك خلال السنة الواحدة، سواء أكانت هذه الإجازات متصلة أم متقطعة، ويقصد بالسنة الواحدة السنة التي تبدأ من تاريخ أول إجازة مرضية.“
وبالمقابل، شدد ”الكاظم“ على أن المصابين يحتاجون إلى تشريعات تحمي حقوق صاحب العمل وتضمن استمرار محارب المرض، وقال: ”ربما هذه الضمانات قد تخيف المؤسسات الرسمية في بداية الأمر، لكن في حيث الشمولية ستتناقص حاجتنا إليها، فعلى سبيل المثال لكل فرد في البحرين يعمل في القطاع العام 24 يوماً، تم تعديلها لصالح مريض الخلية المنجلية إلى 114 يوماً مدفوعة الأجر سنويا، وقد تمكنا بذلك إلى صنع الأمان أن لا أحد يستقطع من راتب المريض ولا أحد يفصله من عمله، فلما اطمأن لم يستخدم الرصيد 24 يوماً فضلا عن الرصيد الإضافي، وأجد أن مجتمع الخلية المنجلية مجتمع مثابر ويكره الشفقة، وإذا أعطيته فرصة يحلق بك إلى السماء“.
وأضاف: ”لا يجوز أن أطالب أصحاب الأعمال وحدهم أن يتحملوا المسؤولية... بل هذه مسؤولية جماعية ومشتركة بين الجهات الرسمية والأهلية والمجتمع الاقتصادي والتشريعي ومجتمع المريض“.
”أحمد“ - 36 سنة - مصاب بنسبة 82% اكتشفت أسرته إصابته بالمرض حين كان في الصف الثالث ابتدائي: ”منذ بداية معاناتي مع المرض كان أول ما تأثر هو مستواي الدراسي ودرجاتي العلمية، ويعود ذلك إلى دخولي فترات طويلة في المستشفى والتنويم لتلقي الرعاية الصحية، تخرجت رغم كل ذلك“.
وقال يصف أول مشواره نحو الوظيفة: ”جاءت المشقة في أول وظيفة لي في مدينة الجبيل التي تبعد عن محافظة القطيف بما يقارب 80 كيلومتراً، ولم تطل مدة عملي فيها، إذ كانت قيادة السيارة كل هذه المسافة مجهدة جداً، في الذهاب والإياب لها بشكل يومي“.
وأكمل يصف حجم ألمه: ”طول مدة الجلوس على مقعد السيارة بشكل ثابت، وجهد التركيز في الطريق يتطلب تحملاً كبيراً لا تطيقه أجسام مصابي فقر الدم المنجلي، فأنا أقود مدة ساعة ونصف في الذهاب ومثلها في العودة، على الرغم من محاولاتي لطلب النقل منها لفرع أقرب لمحافظة القطيف أو داخلها، إلا أنني لم ألق تجاوباً... فاستقلت“.
وراح ”أحمد“ يبحث عن وظيفة أخرى، ولكن كانت تدريباً على حرفة لحام الحديد المنتهية بالوظيفة: ”أثناء التقديم عليها كانت إحدى المتطلبات عمل فحوصات طبية، حيث تم إرفاق تقرير يؤكد وضعي الصحي مع“ المنجلية ”، رغم محاولاتي لإخفاء مرضي لعظم حاجتي لهذه الوظيفة، وعلى ذلك تم إقصائي من البرنامج التدريبي قبل أن يبدأ، لما تتطلبه هذه الوظيفة من جهد جسدي“.
وفي هذه الأثناء، كان ”أحمد“ عاطلاً عن العمل، حين عرف بالإعانة المالية التي تصرف لمصابي فقر الدم المنجلي، خاصة الذين تكون نسبة الإصابة لديهم أكثر من 70%، حيث يصنفون كذوي إعاقة، وتصرف من وزارة الموارد البشرية والتنمية الاجتماعية، عن طريق مركز التأهيل الشامل، إلى جانب 142 مرضاً مزمناً وإعاقة.
وتنقل ”أحمد“ بين عدد من الوظائف طوال سنوات سعيه للعمل، وكل وظيفة لا تستمر أكثر من 9 أشهر، بسبب التعسف الداخلي، في حين كانت تسعفه خططه المالية في الادخار، ولكن المعاناة الحقيقية كانت بعد أن تزوج وأنجب.
و”أحمد“ خريج جامعي بتخصص علوم إدارية، وجد وظيفته التي تتوافق مع تخصصه في إحدى الشركات جيدة المستوى، إلا أن نوبات الألم التي كانت تطرحه الفراش شهراً كاملاً يضطر فيها للتغيب عن العمل، عرضته إلا قرار النقل؛ بسبب تكرر وطول فترات الغياب، حد أن تقترح عليه ذات الشركة للعمل كنادل في أحد المقاهي.
واضطر ”أحمد“ لقبول العمل كنادل مقهى، والتي كانت هي الأخرى مجهدة، وتتطلب الوقوف طوال فترة ساعات الدوام، والتي لم يشفع فيها مراعاة وضعه الصحي، رغم تزويده إياهم تقريراً طبياً يثبت حالته، يطلب فيه السماح له بالجلوس بين ساعات العمل أو تقليل ساعات العمل، ولم يكمل فيها ستة أشهر حتى تعرض للفصل منها هي الأخرى.
وعن تجربة مملكة البحرين في السيطرة على المرض، يتحدث ”الكاظم“ الذي يتقلد أيضا منصب رئيس جمعية البحرين لرعاية مرضى السكلر بأمل كبير معتبراً تجربتها ”نموذج أن المستحيل ممكن“، ويعلل بذلك أن البحرين كانت قبل برنامج الفحص ما قبل الزواج لديها ولادات عالية 4 لكل 100 مولود وخلال 4 سنوات صار 4 لكل 1000، ثم بعد 4 سنوات أخرى صار 6 لكل 10 الآلاف شخص، مشيداً: ”هذا تطور متميز“.
ولفت إلى أن متوسط العمر كان للمصاب 18 عاما ثم أصبح 42 عاماً: ”أما الآن نتحدث عن أعمار تمتد إلى 65 عاماً، ونريد من الفرد الذي يعيش مع هذا المرض أن يموت لأسباب طبيعية كما يتوفى أي شخص بعد أن أتم واجباته ومسؤولياته وقام برسالته في الحياة، ولا نريد طيف الموت يحلق حول رؤوسهم“.
وأوضح أن تجربة مملكة البحرين في رفع متوسط العمر للمصاب لم تحدث، بل جاء ذلك نتيجة إصرار وفهم ووضوح الطريق، وكنا نعلم أين نحن ذاهبون.
وقال: ”حققنا انخفاضاً في الوفيات إلى 62% في العام، والجميع شارك في صناعة النجاح، من مقدمي الرعاية، متلقي الرعاية والمجتمع، الأهل، وصانعي القرار، وواضعي التشريعات والقوانين، صناع الدواء، دور البحث، الجميع شارك حتى الجدة البحرينية شاركت في تشكيل لوبي داخل البيوت لمنع هذه الزيجات بين المصابين“.
وعن إمكانية تحسين جودة حياة الفرد وإطالة عمره، ذكر: "لابد من النظر إلى تجربة البحرين على أنها قصة نجاح يمكن نسخها في أي مكان، فالعيادات متعددة التخصصات واستمرار التدريب والابتعاد عن الوصم بالإدمان وكسب ثقة المريض، فلا يجب أن يكون بين مقدمي الرعاية خلاف مستمر حول طرف يشكو الألم وطرف يتهمه بالإدمان، يجب أن نزيد من فهمنا لهذا المرض ويجب أن نكون أقرب للمريض من أي وقت مضى.
وأكد: ”لا يمكن ذلك بالدواء وحده أو من خلال وجهات نظر طبية فقط، بل من خلال نظرة شمولية، الدراسات مهمة والبيئة العلاجية حيث يسود الحب والتقدير مهم، قد يكلفنا ذاك 10 سنوات، ولكن أؤكد أننا سنتخلص من ما يزيد عن 80% من التحديات اليومية“.
الكاظم الذي يمثل دولياً 27 مليون فرد يعيش مع الخلية المنجلية، يجد أن النظم السياسية التي تجمع الخليج هي ”نظام أبوي“ ينتهج الرعاية والاهتمام، ويتعامل مع الشعب كما لو أنهم ”عياله“، ويخطط ويبادر ويخصص الأموال للتحسين والتطوير.
وأوضح الكاظم بقوله: ”منذ اكتشاف المرض وتسميته امتد إلى أكثر من 100 عام، لم تنتج شركات الدواء أي أدوية لهذا المرض، وما يتوفر هو دواء منذ 25 عاما هو الهيدروكسوريا، وقد شهد العالم دخول 4 أدوية جديدة وهناك عدد واعد منها، وبالنظر إلى الكلفة الشرائية للدواء، سنعرف أن الدواء صنع لدول الخليج فقط، حيث الحكومات هي التي ترعى الدواء، وتوفره مجانا لمواطنيها وحتى للمقيمين فيها“.
واختتم حديثه متألماً: ”في هذه الأثناء من وفرة الدواء وجودة الحياة يصارع الأب في أفريقيا بين أن يشتري الدواء لطفله أو أن يشتري الطعام لـ 18 فرداً يعيلهم، فيفوز الطعام ويموت الطفل. أنا أبكي كثيراً من عجزي أمام فقر القارة الأفريقية ومختلف أنواع الفساد فيها حيث يموت 75% من أطفالها قبل أن يصلوا لعمر 5 سنوات“.
بينما تعاني قارة أفريقيا من ضعف الرعاية الصحية أمام 5 ملايين من حاملي سمة الأنيميا المنجلية، وانخفاض شديد في متوسط عمر الوفاة إلى ما دون الخمس سنوات، يرتفع المتوسط في الولايات المتحدة الأمريكية لذوي العرق الأفريقي من حاملي السمة إلى 58 عاماً، حيث عدد الحاملين فيها يصل إلى 100 ألف شخص، في حين أن متوسط عمر الوفاة في محافظة القطيف دون 30 عاماً.
وطالب بإجراءات أكثر صرامة في سبيل منع إتمام الزواج غير المتوافق طبيًا، وذلك من خلال أخصائيي الوراثة والشريعة، منوهاً بقوله: ”نحن بحاجة لوضع نظام يلزم الأسر ومأذوني الأنكحة باتباع إرشادات الطب الوراثي وتجنب خطورة الإقدام على إتمام مراسم الزواج في حال كشفت التحاليل خطورة ذلك“.
واقترح الدكتور ”رهبيني“ إنشاء ”سجل وطني“ للأمراض الوراثية على غرار ”السجل الوطني للأورام“ وعلل: ”إن إنشاء مثل هذا السجل سوف يوثق الإحصاءات الدقيقة، والتي على أساسها تبنى الاستراتيجيات الوطنية الواضحة، وبالتالي دقة المعايير المستخدمة في فحوصات الأمراض الوراثية وعدم الهدر المالي، وأن تكون أهلية العلاج في هذا المركز متاحة لجميع الحالات من شتى أنحاء المملكة“.
وشجع ”رهبيني“ الأجيال الطبية الشابة على التخصص في مجال الطب الوراثي لمستجدات هذا العلم المتطورة وحاجة الوطن لمثل هذا التخصص سواء من أطباء أو مرشدين وراثيين أو أخصائي تغذية لأمراض التمثيل الغذائي أو باحثين في هذا المجال، وكذلك الحرص على زيادة عدد الأكاديميين في الجامعات لتدريس هذا العلم المتسارع والمتداخل مع العلوم الطبية الأخرى.
وأكد على دور الجمعيات العلمية والتعاون فيما بينها وبين مراكز الأبحاث، والتي تثري النقاش العلمي والبحثي مستقبلا وفتح المجال بين الأطباء والباحثين الشباب، وكذلك زيادة المراجع الطبية المتخصصة في الطب الوراثي لتشمل الوقاية بعد الزواج والفحص أثناء الحمل وتقنية طفل الأنابيب، والتي تعرف بفحص ما قبل الغرس لتشخيص الأمراض الجينية واستبعاد الأجنة المصابة وزراعة الأجنة السليمة داخل رحم الأم ومتابعة الحمل إلى نهايته.
كما لفت إلى أن المملكة تعمل منذ 2013 على بناء قاعدة بيانات وراثية وجينية للمجتمع عبر إطلاق مشروع ”الجينوم السعودي“ للحد من الأمراض الوراثية، وتبلغ تكلفة علاج الأمراض الوراثية في السعودية نحواً مكلفاً، لذا كان من الضروري البحث عن حل جذري للكشف المبكر عنها، والحد من تفاقمها.